
The Hallmarks of Cancer: 2 - Insensitivity to Antigrowth Signals
The Hallmarks of Cancer are ten underlying principles shared by all cancers. You can read the first Hallmark of Cancer article here. The Second Hallmark of Cancer is defined as “Insensitivity to Antigrowth Signals”. Before I explain how failure to respond to antigrowth signals is closely involved in the development of cancer, it is useful to define and understand how cell division works. Cancer is, after all, the uncontrolled division of the cell; so we first need to understand how normal cell division is controlled through the cell cycle.
The Cell Cycle
Think of the Cell Cycle as the control system of a washing machine. A washing machine passes through several stages in a wash cycle; soaking the clothes, adding detergent at the correct time, rinsing the clothes for the appropriate duration to remove the detergent, adding the fabric softener at the correct time, a final rinse and then spinning the clothes to remove as much water as possible. In much the same way, the cell cycle is a series of tightly regulated events inside a cell that lead to its division into two daughter cells. In between these start and end states, the DNA inside the parent cell needs to double and then be divided equally between the two daughter cells. Intricate feedback loops of responsive proteins trigger events in the cell cycle, guiding the cell through checkpoints that lie between every stage. These checkpoints are important because they act as safety valves, ensuring that an errant, incorrectly dividing cell with damaged DNA is promptly whisked away and destroyed rather than allowed to continue its life.
The Four Stages
The cell cycle has four stages.
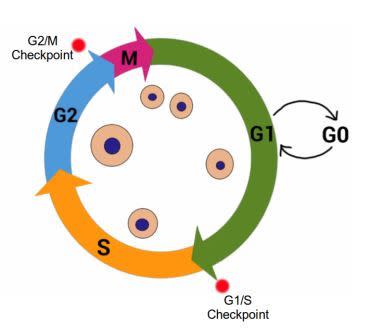
The Cell Cycle, showing G1, S, G2 and M stages. Cell growth occurs during G1 and G2, while DNA is synthesized in S stage. Cell division occurs during M stage (mitosis). G0 indicates quiescent stage where the cell exits the cell cycle but can re-enter if the signals from the environment are appropriate. Red dots indicate important Cell Cycle Checkpoints G1/S and G2/M. Image credit: Buddhini Samarasinghe
Gap 1 (G 1) – during this stage the cell is actively growing in size, and preparing the required components for DNA synthesis. During and at the end of this stage, the cell monitors its surrounding environment (the microenvironment) to make sure there aren’t any ‘stop!!’ signals. This is known as the G1/S Checkpoint.
Synthesis (S) – DNA replication takes place during this stage. If there is damaged DNA in the cell, it should not be replicated, and the G1/S checkpoint ensures this. The G1/S Checkpoint is extremely important to prevent the replication of damaged DNA.
Gap 2 (G2) – The cell continues to grow in the G2 stage following DNA replication. At this stage, it is vital to recognize any damaged DNA before proceeding with cell division. This constitutes the G2/M Checkpoint. If the cell detects damage to its DNA, it will not proceed to the fourth and final stage of the cell cycle. The first G1/S Checkpoint ensures that the template DNA prior to replication is undamaged, and the G2/M Checkpoint ensures that the newly replicated DNA is error-free before proceeding with cell division.
Mitosis (M) – This stage represents the culmination of the cell cycle. Cell growth stops and the cell divides into two equal daughter cells.
Some cells enter a G0 stage known as quiescence which can be considered a place of rest. A cell in G0 has exited the cell cycle, and is neither dividing nor preparing to divide; however the cell is still alive and actively metabolizing, it has simply stopped dividing. The cell may re-emerge from this quiescent stage back into the cell cycle, given the right signals from its microenvironment. Some cells permanently exit the cell cycle, moving to a post-mitotic state. There is no coming back from this path; it is usually associated with mature cells that have differentiated, i.e. taken their adult form. Therefore a cell, at the basic level, has three choices facing it; continue to grow and divide by staying in the cell cycle, take a temporary break by entering G0, or permanently exit the cell cycle into the post-mitotic state.
Controlling the Cell Cycle: Cyclins and CDKs
Two key classes of regulatory proteins control the checkpoints within the cell cycle. These proteins are known as Cyclins and Cyclin Dependent Kinases (CDKs). Cyclins and CDKs cannot work without each other; they need to ‘team up’. Cyclins provide the regulation for the team, and CDKs are the catalyst. CDKs are active only with their specific partner cyclins; this is why they are known as cyclin dependent kinases. In the previous Hallmark of Cancer article, I explained that many proteins exist in an ‘active’ or ‘inactive’ state that can be switched by phosphorylation (i.e. adding a phosphate group to the protein), and that kinases are enzymes that add such phosphate groups to proteins.
A 3D schematic representation of Cyclin A protein. This protein partners with CDK2, for S and G2 stages of the Cell Cycle. Image credit: Wikimedia Commons.
Each stage of the cell cycle has a specific cyclin/CDK pair that behaves as a single unit, called a complex. Each stage’s cyclin/CDK complex is required to allow the cell to commit to and proceed to the next stage of the cell cycle. When activated by a cyclin, a CDK is able to phosphorylate key proteins involved in the networks of chemical reactions in the cell, which in turn are activated and proceed to manufacture the cyclin required for proceeding into the next stage of the cell cycle. It is a beautifully elegant system of control, and it works to ensure that cells grow and divide when they’re supposed to, and remain quiescent at all other times.
Antigrowth signals are proteins, exactly like the growth factors that I mentioned in the previous Hallmarks of Cancer article, except that they inhibit growth rather than promote growth. Antigrowth factors can therefore block cell growth by the two mechanisms mentioned above; cellular quiescence through G0 and the post-mitotic state. A cancer cell must therefore evade these signals if it is to continue dividing uncontrollably. During the G1/S Checkpoint, cells monitor their microenvironment to choose whether to continue in the cell cycle, enter G0 or enter a post-mitotic state. At the molecular level, nearly all antigrowth signals are funneled through a protein known as the Retinoblastoma protein. Therefore, Retinoblastoma is classified as a tumor suppressor protein. Indeed, it was the very first tumor suppressor protein to be discovered in 1971 through an elegant process of deduction and statistical analysis of rare eye cancers.
Retinoblastoma Protein
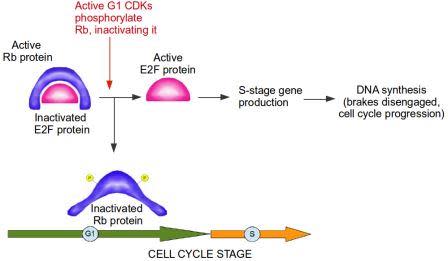
If a cell detects that it has damaged DNA at the G1/S checkpoint, this damaged DNA should not be replicated. Therefore, at the G1/S checkpoint, prior to entry into S stage, the brakes are slammed and everything comes to a screeching halt. In this analogy, the Retinoblastoma protein acts as the brake. Retinoblastoma protein is active when it is not phosphorylated, meaning phosphorylation inactivates Retinoblastoma. A primary function of Retinoblastoma is to bind to and inactivate E2F transcription factors. These are extremely important proteins that bind to DNA and activate genes which…you guessed it, control the cell cycle and DNA replication, including the Cyclins and CDKs specific to G1 and S phases of the cell cycle. In other words, E2F transcription factors are controlled by their interaction with the Retinoblastoma protein, which acts as the brake in the cell cycle progression to the S Stage. During G1, Retinoblastoma protein binds to E2F and blocks the production of S stage Cyclins and CDKs. When cells are stimulated to divide by external signals in their microenvironment, active CDKs specific to the G1 stage accumulate and phosphorylate Retinoblastoma. The Retinoblastoma protein, now inactivated, moves away from E2F, allowing the cell cycle to proceed. The brake is disengaged (see diagram below).
Active Rb (Retinoblastoma protein) inactivates E2F protein. CDKs (cyclin-dependent kinases) from G1 stage of the cell cycle phosphorylate Rb, thereby inactivating it. E2F is now active and free to produce genes required for progression of cell cycle into S stage. The brake, from Rb, is disengaged. Image credit: Buddhini Samarasinghe
Without E2F transcription factors, cell division grinds to a halt, and the phosphorylation status of Retinoblastoma controls the activity of these E2F transcription factors! Think about it for a moment: the addition or removal of a tiny molecule of phosphate to a single Retinoblastoma protein within the cell is responsible for whether a cell divides or not!
What influences the activation (i.e. phosphorylation status) of the Retinoblastoma protein? Antigrowth signals from the surrounding microenvironment of the cell. Given how important a role Retinoblastoma plays in the control of cell division, it comes as no surprise that cancer cells have found a myriad ways to bypass these antigrowth signals. Disruption of the retinoblastoma pathway liberates E2F transcription factors to promote cell division, and thus cells become insensitive to antigrowth signals that normally control this process.
Antigrowth signals
So what are these antigrowth signals? Probably the best documented is the signaling molecule TGF-beta. Recall from the previous paragraph that phosphorylated Retinoblastoma is inactive, and this ‘disengages’ the brakes on the cell cycle. TGF-beta has many different mechanisms for preventing the phosphorylation of Retinoblastoma (i.e. for preventing the disengagement of the brakes). Therefore, the presence of TGF-beta blocks the advancement of the cell cycle. Unsurprisingly, many cancers target this pathway for disruption. Some cancer cells stop responding to TGF-beta altogether, by producing less TGF-beta receptors on their cell surfaces. Other cancers produce mutated receptors that do not respond to the presence of TGF-beta. Some cancers get rid of downstream proteins that respond to TGF-beta. In many late-stage tumors, instead of functioning as an antigrowth signal, TGF-beta activates a cellular program known as Epithelial-to-Mesenchymal-Transition (EMT), which gives cancer cells stem-cell like abilities that is really bad news to a cancer patient. Finally, Retinoblastoma protein itself, the end target of this pathway, can be lost through mutation of its gene. Interestingly, certain cancer-promoting proteins (oncoproteins) can block the function of Retinoblastoma as well. For example, the human papillomavirus produces a protein known as E7, which binds to and inactivates Retinoblastoma.
The end result is that antigrowth signals, funneled through Retinoblastoma protein into the cell cycle, are, in one way or the other, disrupted in a majority of human cancers. Cancer cells with defects in the Retinoblastoma pathway are missing the services of a critical ‘gatekeeper’ of cell cycle progression; the absence of the Retinoblastoma gatekeeper permits persistent cell division. Therefore, the insensitivity to antigrowth signals represents a vital breach of an anti-cancer defense mechanism that is hardwired into our cells.
Next time…”Evading Apoptosis”
Follow Scientific American on Twitter @SciAm and @SciamBlogs. Visit ScientificAmerican.com for the latest in science, health and technology news.
© 2013 ScientificAmerican.com. All rights reserved.
