
Hallmarks of Cancer 3: Evading Apoptosis



The Hallmarks of Cancer are ten underlying principles shared by all cancers. The first and second Hallmark of Cancer articles can be found here and here.
Apoptosis is also observed in normal embryonic development, not just for cancer. On the left is the paw of a mouse embryo, 15 days after fertilization. On the right is an enlarged view of the same paw, showing enhanced apoptosis in cells between the webbing of the fingers (the glowing green dots are apoptotic cells). Image credit: Amitabha Bandyopadhyay et. al., 2006 PLOS Genetics doi:10.1371/journal.pgen.0020216 (http://goo.gl/vkdaQA)
The Third Hallmark of Cancer is defined as “Evading Apoptosis”. Apoptosis is the opposite of cell growth; it is cell death. To divide and grow uncontrollably, a cancer cell not only has to hijack normal cellular growth pathways, but also evade cellular death pathways. Indeed, this acquired resistance to apoptosis is characteristic of all types of cancer. But before I explain how cancer cells do this, we need to understand how the process of cellular death occurs in a normal cell.
The apoptotic program is hardwired into every single cell in our body. It is like a cyanide capsule, swiftly delivering death if the circumstances require cellular suicide. If a cell detects that it has damaged DNA, it can activate apoptosis to remove itself from the population. Apoptosis, or cellular suicide, is an entirely normal function of cells. The same apoptotic program is activated when a tadpole changes into a frog; the cells in the tail die through apoptosis, and the tail disappears. The same is true for the webbing between our fingers in our early embryonic development. Apoptosis is an extremely tidy process; cellular membranes are disrupted, the chromosomes are degraded, the DNA breaks up into fragments, and the dying, shrinking cell is swallowed up by a neighboring cell or a patrolling immune cell, leaving no trace of the cellular suicide behind.
Regulators and Effectors
So how does apoptosis work at the molecular level? The apoptotic machinery can be divided into two broad categories; regulators and effectors. The regulators are responsible for monitoring the interior and exterior environment of the cell for conditions of abnormality in order to decide whether that cell should live or die. The possible abnormalities include DNA damage, signaling imbalance caused by the activation of cancer causing genes (oncogenes), lack of an oxygen supply or insufficient growth factors.
The Bcl-2 family of proteins can be divided into pro-apoptotic proteins and anti-apoptotic proteins, i.e. proteins that promote apoptosis and proteins that inhibit apoptosis. *For an explanation of the 'interesting' names for the pro-apoptotic proteins, see note at the end of this article! Image credit: Buddhini Samarasinghe
Apoptosis can therefore occur either through an intrinsic pathway, in which signals from within the cell activate the process, or through an extrinsic pathway where death signals from outside the cell are received and processed by the cell to activate apoptosis. It is thought that the intrinsic apoptotic pathway is more important in cancer prevention than the extrinsic pathway. Given how our cells carry machinery to destroy themselves with the precision of an executioner, it comes as no surprise that the process is tightly regulated.
The primary regulators of apoptosis are proteins belonging to a group known as the Bcl-2 family. These proteins can either be pro-apoptotic or anti-apoptotic; Bcl-2, Bcl-XL, Bcl-W, Mcl-1 and A1 proteins function as anti-apoptotic proteins that inhibit apoptosis, while Bax, Bad, Bid, Bok, Bik and Bak (I swear these names are not made up!*) are pro-apoptotic proteins that trigger apoptosis when activated. The anti-apoptotic proteins bind to and inactivate the pro-apoptotic proteins in a healthy cell that does not need to die. Apoptosis regulators also include death receptors on the cell surface which bind to death signaling molecules, as part of the extrinsic apoptotic pathway. This is similar to the way growth factors bind to and activate growth factor receptors, as I described previously, and this binding triggers the effectors of apoptosis.
The Suicide Machines
A brief overview of the extrinsic and intrinsic apoptotic pathways. In the extrinsic pathway, death signals from the surrounding environment of the cell bind to death receptors on the surface of the cell membrane. This causes the conversion of inactive pro-caspase-8 into active caspase-8. Caspase-8 then goes on to activate caspase-3, which begins the caspase cascade that leads to apoptosis. In the intrinsic pathway, typically initiated by DNA damage, P53 is activated. P53 then activates the pro-apoptotic protein Bax, which initiates the release of cytochrome c from the mitochondria. Apaf-1 and the released cytochrome c combine to form a complex known as the apoptosome. The apoptosome causes the conversion of inactive pro-caspase-9 into active caspase-9. Caspase-9 then goes on to activate caspase-3 in a similar manner to the extrinsic pathway. Activated caspase-3 then leads to the caspase cascade, resulting in apoptosis. Image credit: Buddhini Samarasinghe
Where is Mission Control for apoptosis? Many of the apoptotic signaling pathways converge at the mitochondria. Mitochondria are tiny organelles floating within a cell, and function as the cell’s energy factories. They contain a signaling molecule known as cytochrome c, which is bound to the mitochondrial membrane. In response to pro-apoptotic signals (from pro-apoptotic proteins such as Bax), cytochrome c is released into the cell by the mitochondria, and they bind to a protein known as Apaf-1. This results in the formation of the apoptosome. The apoptosome is an extremely beautiful structure resembling a wheel with seven spokes. Once formed, the apoptosome goes on to activate a group of proteins known as caspases.
All our cells contain the seeds of their own destruction; these come in the form of caspases. Caspases can be thought of as cellular executioners. They are proteins that degrade other proteins in our cells. Active caspases can wreak havoc within a cell and are therefore extremely dangerous, so they are produced in an inactive form by the cell (known as pro-caspases), like an executioner’s blade that is sheathed. Upon detecting an increase in the amount of cytochrome c, released from the mitochondria, the blades are unsheathed. There are 13 such caspase genes identified in the human genome so far. Two of the caspase proteins act as ‘gatekeeper’ caspases: caspase-8 and caspase-9. They are initiator caspases that, when activated by cytochrome c release, go on to activate the other caspases in a cascade of irreversible cellular protein degradation.
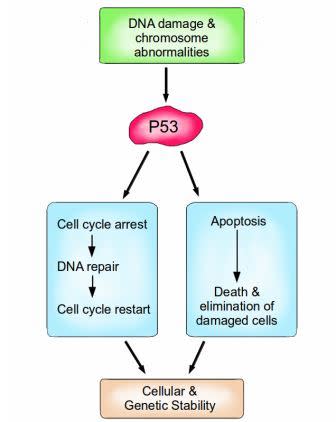
P53: The Guardian of the Genome
P53 is a vital protein; more than half of all cancers have inactive P53. When activated by either DNA damage or chromosome abnormalities, P53 can halt the cell cycle and initiate DNA repair. If repair is successful, the cell cycle is restarted, and cellular and genomic stability is re-established. If repair is not an option because the damage is too great, then P53 can facilitate apoptosis, leading to the death and elimination of the abnormal cell. Image credit: Buddhini Samarasinghe
How do cells detect the necessary conditions for triggering apoptosis? In the previous Hallmark of Cancer article, I explained the fundamental role of the Retinoblastoma protein in controlling cell division. Retinoblastoma, remember, is a vitally important brake on cell division. Damage to a cell’s Retinoblastoma gene releases this brake, leading to uncontrolled cell growth.
Similarly, P53 is an extremely important protein, dubbed ‘The Guardian of the Genome’. Amongst its many functions, it is responsible for detecting DNA damage, chromosome abnormalities and arresting the cell cycle to initiate repair; if repair is not possible then apoptosis is induced. P53 induces apoptosis by increasing the production of the pro-apoptotic protein Bax. Bax stimulates the mitochondria to release cytochrome c, which activates the caspase cascade which ultimately results in cell suicide. P53 is vital for maintaining the integrity of our genome at the most fundamental level.
Evading apoptosis
So how do cancer cells escape death? The most common method is the loss of the apoptosis gatekeeper, the protein P53. More than half of all types of human cancers have a mutated or missing gene for p53, resulting in a damaged or missing P53 protein. As an alternative to achieving the loss of P53, cancer cells can compromise the activity of P53 by increasing the inhibitors of P53, or silencing the activators of P53. Previously I explained how the human papillomavirus produces a protein known as E7, which binds to and inactivates Retinoblastoma.
Similarly, another protein, E6, also produced by the human papilloma virus, binds to and inactivates P53. These two cancer-causing proteins, E6 and E7 (oncoproteins), therefore disable two vital gatekeepers, Retinoblastoma and P53, that control both cell division and cell death; the result is repeated uncontrolled cell division that manifests itself in warts, with strong associations with the development of cancer. Cancer cells can also produce excessive amounts of anti-apoptotic proteins such as Bcl-2, Bcl-XL etc. They can produce less of the pro-apoptotic proteins such as Bax and Bak. They can short-circuit the extrinsic death receptor apoptotic pathway.
It comes as no surprise that highly aggressive cancers often have both Retinoblastoma and P53 mutations. As a result, these rapidly growing tumors have extremely low levels of apoptosis and extremely high levels of cell division. Like de-rigging the Sword of Damocles, cancer cells can inactivate the machineries of death; and the evasion of apoptosis by cancer cells therefore represents a key breach of an extremely important anti-cancer defense mechanism.
Next time…”Limitless Replicative Potential “
*in case you were curious, the pro-apoptotic members of the Bcl-2 family are thus named:
Bax: Bcl-2 Associated X protein
Bad: Bcl-2 Associated Death promoter
Bid: BH3 interacting-domain Death agonist
Bok: Bcl-2 related Ovarian Killer
Bik: Bcl-2 Interacting Killer
Bak: Bcl-2 homologous Antagonist Killer
Follow Scientific American on Twitter @SciAm and @SciamBlogs. Visit ScientificAmerican.com for the latest in science, health and technology news.
© 2013 ScientificAmerican.com. All rights reserved.
