
The Hallmarks of Cancer: 4 - Limitless Replicative Potential

The Hallmarks of Cancer are ten underlying principles shared by all cancers. The previous Hallmark of Cancer articles can be found here. The Fourth Hallmark of Cancer is defined as “Limitless Replicative Potential”.
The first three Hallmarks of Cancer explain how independence from growth signals, insensitivity to antigrowth signals and resistance to apoptosis lead to the uncoupling of a cell’s growth program from the signals in its environment. However, cancer is not just a result of disrupted signaling. Our cells carry an in-built, autonomous program that limits their multiplication, even in the face of disrupted signals from their environment. For a single cancer cell to develop into a visible tumor, this program must also be disrupted.
The Cellular Timekeeper
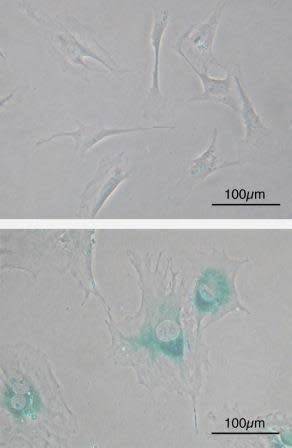
Microscope image of normal cells (upper) compared to senescent cells (lower). Blue-green areas indicate expression of marker associated with senescence. Image credit: Wikimedia Commons.
Normal cells are hard wired with a timer that keeps track of their age; the number of times they divide and grow. Most cells in our body can only undergo a limited number of successive cell growth-and-division cycles. This limit is named the Hayflick Limit after its discoverer, Leonard Hayflick. After undergoing between 40 and 60 divisions, cell growth slows down and eventually stops altogether. This state is known as senescence, and it is irreversible; although the cell does not grow or divide, it remains alive. When normal human cells are cultured in the lab in a petri dish, we can observe this phenomenon, where cells grow and divide a fixed number of times and then enter senescence. Some cells are able to make it past the senescence barrier and continue dividing; however these cells then undergo a second phenomenon known as crisis, during which the ends of their chromosomes fuse with each other, and the cells all die on a massive scale via apoptosis.
How does a cell count its divisions? How does it ‘know’ when to stop? The answer is telomeres. Telomeres are regions of repetitive DNA, capping and protecting the ends of the chromosome from degrading or from fusing with another chromosome. Without telomeres, each time a cell divides our genomes would progressively lose information because the chromosomes would get shorter and shorter. A telomere is like the heat-shield of a spacecraft; it protects the actual spacecraft and absorbs the damage instead. With every replication of a cell, about 50-100 nucleotides of telomeric DNA is lost. This progressive loss eventually causes the telomeres to lose their ability to protect the ends of chromosomal DNA. Left unprotected, these exposed ends become damaged. The DNA damage response is activated, leading to growth arrest; senescence. When chromosome ends fuse with each other, this irreversible damage results in the activation of apoptosis; the cell enters crisis, and dies.
The End Replication Problem
Why do the ends of chromosomes shorten? To understand this, first we need to go over the basic mechanisms of DNA replication. A cell must replicate its DNA before it divides. DNA is a double-stranded molecule, and each strand of the original DNA molecule serves as a template for the production of a complementary strand. The two strands have a directionality; the two ends of a single strand are known as the 3′ end and the 5′ end. The numbers refer to the position of the carbon atom in the deoxyribose molecule at the end of the strand to which the next phosphate molecule in the DNA chain attaches. For a quick introduction into the structure of DNA, check out this YouTube video:
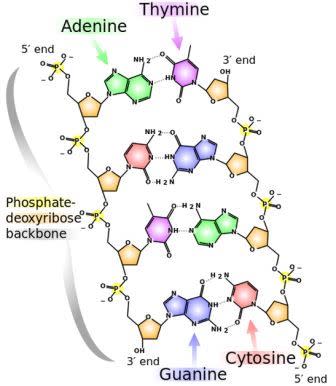
The chemical structure of DNA. The 5' and 3' ends are shown lying antiparallel to each other. Nitrogenous bases adenine (green), thymine (purple), guanine (blue) and cytosine (red). These bases are fixed to the phosphate-deoxyribose backbone. Image credit: Wikimedia Commons.
This directionality matters because DNA replication takes place under the direction of an enzyme known as DNA polymerase. This enzyme faithfully copies our genetic code letter by letter. However, DNA polymerase can only work in one direction; the 5′ to 3′ direction. Therefore, although DNA replication is straightforward for one of the DNA strands (the 5′ to 3′ strand), the other strand (the 3′ to 5′ strand) is more complicated. This strand is replicated in short fragments instead of one continuous strand of DNA. Replication begins with an enzyme known as primase, which reads the template DNA and initiates the synthesis of very short complementary RNA fragments. DNA polymerase is now able to use these RNA fragments as a starting point to synthesize complementary fragments of DNA in between the RNA fragments. The RNA fragments are then removed and replaced with DNA, and the fragments of DNA are joined together by another enzyme, DNA ligase. This solves the directionality problem of DNA polymerase, but now we run into another problem, known as the end replication problem. This is because although the 5′ to 3′ strand can be replicated to the very end, the 3′ to 5′ strand cannot; DNA polymerase enzyme requires RNA fragments to begin replication, and there is nothing for such a fragment to attach to at the very end of the DNA strand. Therefore, with every round of replication, a small fragment of the DNA would be lost from the end of the chromosome, since it cannot be replicated. The cell solves this problem by having telomeres at the ends of chromosomes, where they prevent the loss of valuable genetic information by acting as a disposable buffer. Over time, with each successive round of DNA replication, the telomeric DNA shortens until finally there is no more disposable buffer, at which point the cell stops dividing and enters senescence. For a visualization of the end replication problem check out this YouTube video:
When cells are grown in petri dishes in the lab, repeated cycles of cell division lead first to senescence and then, for those cells that make it past this barrier, to crisis phase. Fascinatingly, in very rare instances (about 1 in 10X7) a cell can emerge from this ordeal exhibiting unlimited replicative potential. This cell is now said to be immortalized, and it is a trait that most cancer cells growing in labs exhibit, including the famous HeLa cells.
Telomere Maintenance
1. Double stranded DNA is made up of two DNA strands lying antiparallel to each other, with their 5' and 3' ends highlighted in red. 2. When DNA replicates, the two strands unwind. Replication of the Leading Strand is very straightforward, with the DNA polymerase enzyme copying from the purple template strand to form the new DNA strand, shown in orange. This new strand is formed in the 5' to 3' direction, since DNA polymerase only works in that direction. 3. For the synthesis of the Lagging Strand, the enzyme RNA Primase creates short complementary RNA fragments, shown in green. 4. DNA polymerase is now able to use these short RNA fragments as a starting point to synthesize DNA between the RNA gaps, shown in orange. The enzyme still works from 5' to 3', in a 'backstitching' manner. 5. The RNA primers are now removed, and the gaps are filled in by DNA polymerase to form a complete replicate of the lagging strand. 6. However, the extreme end of the DNA strand cannot be replicated because there is nowhere for the DNA polymerase enzyme to attach to. 7. The presence of a telomere, shown in pink, allows the DNA to be protected as the telomere gets shortened instead. Image credit: Buddhini Samarasinghe
Cancer cells have therefore not only uncoupled their growth program from the signals in their environment, they have also breached the in-built replication limit hard wired into the cell. How do they achieve this? All cancer cells maintain their telomeres. 90% of them do so by increasing the production of an enzyme known as telomerase. As its name implies, telomerase functions by adding telomeric DNA to the ends of chromosomes. Most normal cells do not divide frequently, and therefore are not in any danger of shortened telomeres; these cells can get away with having low telomerase activity. Indeed, most cells apart from fetal cells and stem cells show low telomerase activity levels. Many cancer causing proteins (oncoproteins) are able to activate the production of telomerase, while many cancer preventing proteins (tumor suppressors) such as P53 (see previous Hallmark) produce factors that inhibit the production of telomerase. The other 10% of cancers rely upon the activation of a pathway known as the Alternative Lengthening of Telomeres (ALT), which swaps around telomeres to lengthen them.
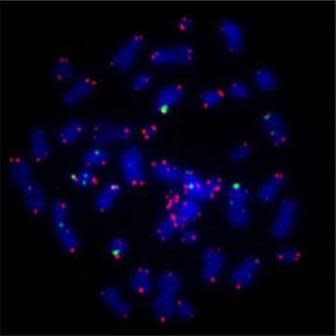
Human chromosomes, stained blue, with marker for telomere region in red. Image credit: Asako J. Nakamura (Wikimedia Commons).
Intriguingly, telomere length is also affected by oxidative stress. Oxidative stress, in the form of free radicals, damages DNA. This damage is usually repaired by DNA repair mechanisms, but these mechanisms are less effective on telomeric DNA than elsewhere on the chromosome. Telomeres are therefore highly susceptible to oxidative stress. It also explains the rate of telomere shortening observed; estimated loss per cell division because of the end replication problem has been estimated at 20 base pairs of DNA, yet the observed loss is much larger, between 50-100 base pairs of DNA. This difference shows that oxidative stress has a far greater impact on telomere length than the nuances of DNA replication. It is possible that cell senescence induced by telomere loss is therefore a stress response, evolved to block the growth and replication of cells that have been exposed to a high risk of DNA damage.
The defining feature of a cancer cell is its ability to divide endlessly, without exhaustion, generation after generation. They achieve this by destroying the cellular timekeeper, the telomere. Immortality comes at a price; the accumulation of damaging mutations only increases with time, which is why cancer is primarily a disease of an aging population. The immortalization of cancer cells by telomere maintenance therefore represents an essential step in tumor progression.
Follow Scientific American on Twitter @SciAm and @SciamBlogs. Visit ScientificAmerican.com for the latest in science, health and technology news.
© 2013 ScientificAmerican.com. All rights reserved.
