India accounted for 1 in 4 coronavirus deaths worldwide last week
India accounted for one in four of the world's recorded coronavirus deaths last week, WHO said.
It said India accounted for 46% of cases and 25% of deaths in the period.
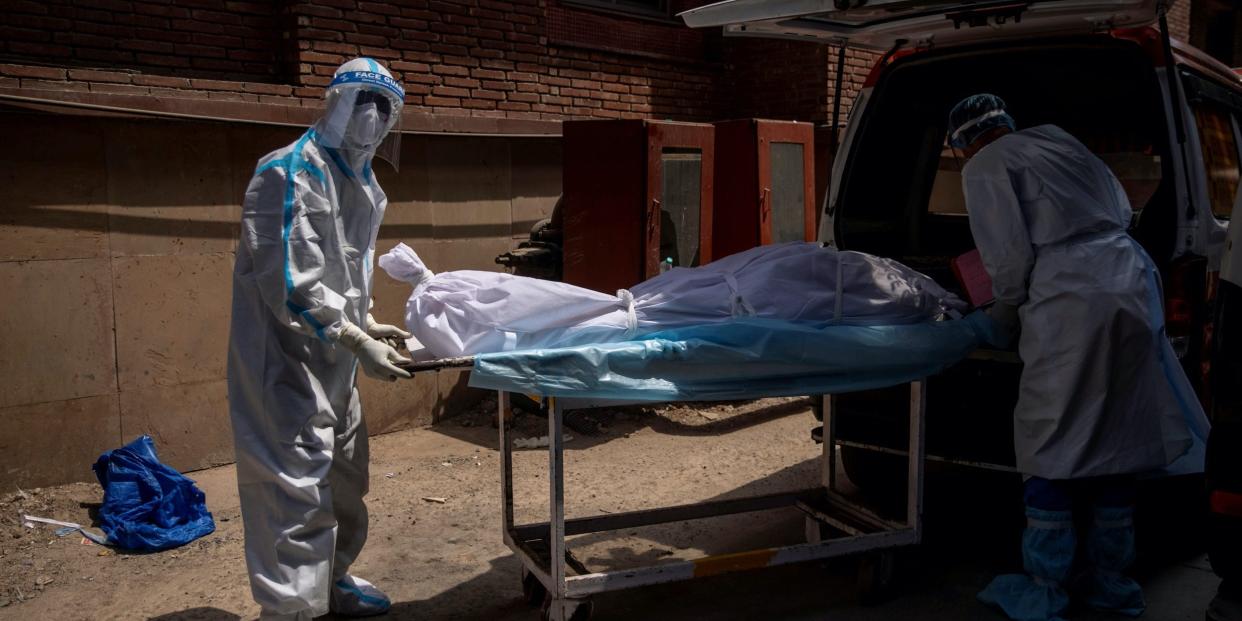
India is facing a devastating surge that has overwhelmed hospitals, morgues, and crematoriums.
India accounted for one in four coronavirus deaths recorded around the world last week, the World Health Organization said.
WHO said in its weekly epidemiological update, published Tuesday, that India recorded 46% of global cases and 25% global deaths from April 26 to May 2.
It said 5.7 million new cases and more than 93,000 deaths were reported around the world last week. Some 2.6 million of those cases were recorded in India, the agency said.
India is facing a record-breaking surge of the virus that has overwhelmed hospitals, morgues, and crematoriums. Hospitals are struggling with a critical oxygen shortage.
As of Wednesday, the country had recorded more than 20 million coronavirus cases and more then 226,000 deaths, according to data from Johns Hopkins University.
It also reported a record 3,780 deaths in the 24 hours before Wednesday.
Experts have said India's true COVID-19 death toll could be up to 10 times the known figure.
Read the original article on Business Insider

